 Fraunhofer-Institut für Verfahrenstechnik und Verpackung IVV
Fraunhofer-Institut für Verfahrenstechnik und Verpackung IVVIm Folgenden werden speziell nur Gerätetypen dargestellt, die im Fraunhofer IVV zur Verfügung stehen. Auf TOF-MS und HPLC-MS wird in diesem Dokument nicht eingegegangen.
1 Grundlagen der Massenspektrometrie
2 Quadrupolmassenspektrometer
2.1 Probenaufgabe
2.2 Ionisierung der Probe
2.3 Analysator
2.4 Detektor
3 Hochauflösende Sektorfeldmassenspektrometer (HRMS)
4 Ion Trap MS-Systeme
4.1 Der Aufbau des Systems
4.2 Die Bestandteile der Ion Trap
4.3 Zusammenfassung der möglichen Ion Trap Parameter
4.3.1 Steuerung der hochfrequenten Wechselspannung
4.3.2 Axiale Modulationsspannung
4.3.3 Gasdruck in der Ion Trap
4.3.4 Elektronenstoßionisation mit Automatic Gain Control
4.4 Geräteoptimierung am Ion Trap
5 Unterschiede im Auflösungsvermögen verschiedener Massenspektrometertypen
5.1 Allgemeine Definition des Auflösungsvermögens
5.2 Auflösungsvermögen von Sektorfeldmassenspektrometern
5.3 Auflösungsvermögen von Quadrupolmassenspektrometern
5.4 Auflösungsvermögen von Ion Trap Massenspektrometern
Literaturverzeichnis
1 Grundlagen der Massenspektrometrie
Die Funktionsweise eines Massenspektrometers (MS) kann wie folgt skizziert werden: Die zu untersuchende Probe wird in das MS eingebracht, verdampft und ionisiert. Als bewegte geladene Teilchen lassen sich die Ionen in einem Analysator auf verschiedene Weise nach ihrem Masse-zu-Ladungs-Verhältnis auftrennen und anschließend detektieren. Der Aufbau eines MS läßt sich somit in vier Hauptkomponenten aufgliedern: Probenaufgabesystem, Ionisierung, Massentrennung und Detektion (Abb. 1).
Die technische Realisierung von Probenaufgabe, Ionisierung und Detektion ist bei den im Folgenden gezeigten Quadrupol- und Sektorfeldgeräten vergleichbar. Das in Kapitel 4 vorgestellte Ion Trap System unterscheidet sich dahingehend, dass es sich um ein Speichermassenspektrometer mit Ionisierung in der Trap handelt.
Die Probenaufgabe in Massenspektrometer erfolgt je nach Eigenschaften der Probe. Feste Probensubstanzen können direkt über eine Schubstange in die Ionenquelle eingebracht werden. Für flüssige oder gasförmige Proben eignet sich die Kopplung mit einem Gaschromatographen (GC) oder Hochdruckflüssigkeitschromatographen (HPLC). Der wesentliche Unterschied besteht in den Analysatorsystemen, die für die Massentrennung zuständig sind.
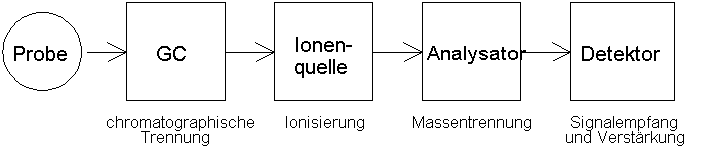
Abb. 1: Schematischer Aufbau eines GC/MS-Systems
2 Quadrupolmassenspektrometer
Normalerweise erfolgt die Probenaufgabe über einen Gaschromatographen, der über ein Interface direkt mit dem MS gekoppelt ist. Eine hochauflösende Kapillarsäule trennt die Dioxin- und Furanmoleküle nach Polarität und Größe der Moleküle auf. Über das Interface gelangen die Fraktionen in die Ionisierungskammer. In dieser Kammer ist ein Vakuum von ca. 10-6 mbar erforderlich, welches von einer Turbomolekularpumpe hergestellt wird. Die Leistung der im QP-5000 standardmäßig eingebauten Pumpe (50 l/s) limitiert den Innendurchmesser der Kapillarsäule auf max. 0,32 mm, eine am QP-5000 mit Pyrolysator eingebaute größere Pumpe hat eine Pumpleistung von 150 l/s und erlaubt auch größere Säuleninnendurchmesser.
Es gibt in der Massenspektrometrie unterschiedliche Ionisierungstechniken. Das am Fraunhofer IVV verwendete QP-5000 ist in der Standardausführung nur für Elektronenstoßionisation (EI) ausgelegt. Die Probemoleküle gelangen aus dem Interface direkt in die Ionisationskammer. Dort befinden sich, einander gegenüber plaziert, die sogenannten Filamente, zwei dünne Drähte aus einer Rheniumlegierung. Hinter den Drähten ist jeweils eine Metallplatte angebracht. Eines der beiden Filamente dient als Elektronenquelle, in dem man es elektrisch weißglühend aufheizt, so dass durch Thermoemission Elektronen frei werden. Zwischen dem Filament und der Metallplatte auf der gegenüberliegenden Seite liegt eine Spannungsdifferenz von 70 Volt an, die die Elektronen durch den Ionisierungsraum beschleunigt. Sie stoßen mit den Probemolekülen zusammen und schlagen durch den Stoß weitere Elektronen heraus. Dabei entstehen größtenteils positiv geladene Ionen mit einfacher Ladung nach folgendem Muster:
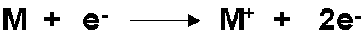
Negative Ionen entstehen bei EI mit etwa 1000 mal geringerer Wahrscheinlichkeit. Da die Energie der Elektronen 70 eV beträgt, zur Ionisierung der Probemoleküle jedoch nur etwa 5-20 eV benötigt werden, können die Moleküle durch die überschüssige Energie an labilen Bindungen auseinander brechen. Das Fragmentierungsmuster ist typisch für eine bestimmte Substanz und damit ein wichtiger Hinweis bei der Identifizierung. Der Betrag von 70 eV Ionisierungsenergie wird bevorzugt gewählt, weil die Ionenausbeutekurve für organische Moleküle in diesem Bereich ein Maximum durchläuft und zu dem so flach ist, dass gute Reproduzierbarkeit der Spektren auch bei geringen Schwankungen der Ionisierungsenergie gegeben ist.
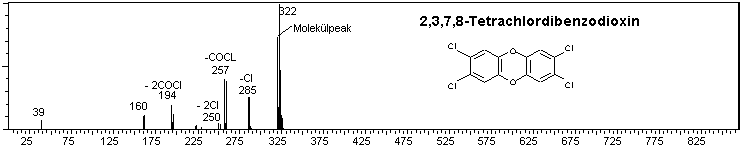
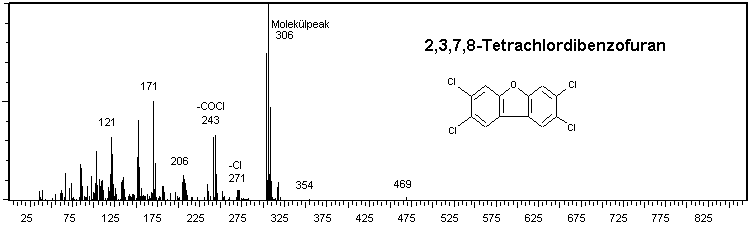
Abb. 2: EI-Massenspektren von 2,3,7,8-TCDD und 2,3,7,8-TCDF
Die gebildeten Ionen werden durch ein kleines Ziehpotential an einer Extraktionsblende aus der Ionenkammer heraus in Richtung des Analysators beschleunigt. Sie passieren dann eine Reihe von Linsen, deren Aufgabe es ist, diesen Ionenstrahl möglichst scharf zu fokussieren.
Der Analysator besteht aus vier zylindrischen parallel angeordneten Stäben, von denen jeweils die sich gegenüberliegenden Stäbe elektrisch miteinander verbunden sind. Ein ideales gleichmäßiges elektrisches Quadrupolfeld würde sich ausbilden, wenn die Stäbe Hyperbelform aufweisen. Aus fertigungstechnischen Gründen sind die Stäbe aber beim QP-5000 zylindrisch geformt.
Für die Detektion positiver Ionen liegt die Ionisierungskammer gegenüber dem Quadrupol auf einem positiven Potential. Diese Potentialdifferenz bestimmt die kinetische Energie der Ionen, mit der sie den Analysator durchfliegen. Auf jedem Stabpaar liegt eine Gleichspannung Vdc (ca. 0,5 kV) an, die von einer Wechselspannung VRF (ca. 6 kV) mit einer Frequenz von 1,2 MHz überlagert ist, wobei die Stabpaare entgegengesetzte Polarität haben (Abb. 3).
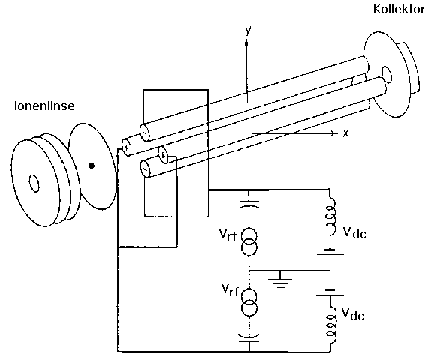
Abb. 3: Quadrupolanalysator
Das elektrische Quadrupolfeld, das sich ausbildet, kann mit den Mathieuschen Differentialgleichungen beschrieben und berechnet werden. Aus den Lösungen dieser Differentialgleichungen folgt, dass man die Spannungen an den Quadrupolstäben derart einstellen kann, dass nur Ionen mit einem bestimmten Masse-zu-Ladungsverhältnis stabile Bahnen auf ihrem Weg durch den Analysator beschreiben und den Detektor erreichen. Alle anderen Ionen bewegen sich auf instabilen Bahnen. Ihre Amplitude wächst exponentiell an, so dass sie an die Quadrupolstäbe geraten und dort entladen werden. Im Gegensatz zu anderen Massenspektrometerbauarten kann das Quadrupolgerät damit als echtes Massenfilter bezeichnet werden. Zur Massenselektion variiert man gleichzeitig die Gleichspannung und die Amplitude der Wechselspannung in einem bestimmten Verhältnis; die Frequenz bleibt konstant (Abb. 4).
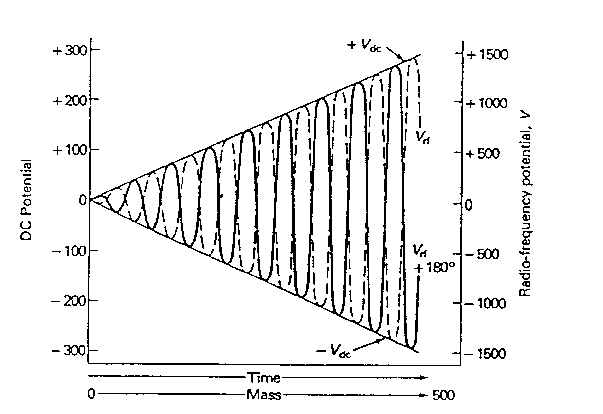
Abb. 4: Zeitlicher Verlauf von Gleichspannung Vdc und Wechselspannung VRF*coswt beim Durchfahren eines Massenspektrums
Das QP-5000 verfügt zusätzlich über Vorstäbe vor jedem der vier Hauptstäbe. Sie sind von den Hauptstäben elektrisch isoliert und werden nur mit einer Wechselspannung versorgt. Durch sie können die Ionen von der Ionenquelle aus das Randfeld an den Enden des Quadrupols durchdringen und werden besser zur Zentralachse des Quadrupol fokussiert. Außerdem werden die Hauptstäbe sauberer gehalten, weil nichtselektierte Ionen von der Zentralachse ablenkt werden und nicht in den Analysator gelangen.
Als Detektor dient ein Sekundärelektronenvervielfacher (SEV). Die Ionen treffen hier auf die Oberfläche einer Dynode und schlagen Elektronen aus ihr heraus. Diese Elektronen treffen durch den kaskadenförmigen Aufbau des SEV auf die nächste Dynode und schlagen ihrerseits weitere Elektronen heraus. Dadurch wird eine Signalverstärkung bis zu 106 erreicht. Der kaskadenförmige Aufbau ist beim QP-5000 durch eine Halbleiterschicht realisiert worden. Am Ausgang des SEV ist ein Elektrometer-Verstärker nachgeschaltet, der den Strom in eine Spannung umwandelt, die als Messgröße weiterverarbeitet werden kann.
3 Hochauflösende Sektorfeldmassenspektrometer (HRMS)
Sektorfeld- und Quadrupolmassenspektrometer sind im Wesentlichen aus den gleichen Komponenten aufgebaut. Die Funktionsweise dieser Komponenten ist jeweils vergleichbar. Der hauptsächliche Unterschied zwischen den beiden Geräten ist das Analysatorsystem, das daher im folgenden für das HRMS genauer beschrieben wird. Kernstück eines hochauflösenden Sektorfeldmassenspektrometers ist die doppelte Fokussierung des Ionenstrahls durch ein Magnetfeld und ein elektrisches Feld, die nacheinander angeordnet sind. Auch die umgekehrte Reihenfolge ist möglich (Abb. 5).
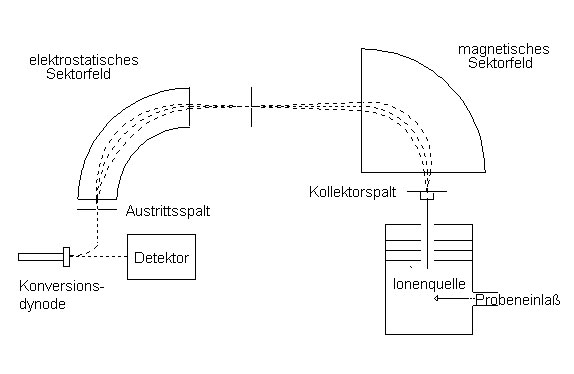 Abb. 5: Analysatorsystem eines doppeltfokussierenden Massenspektrometers
Abb. 5: Analysatorsystem eines doppeltfokussierenden Massenspektrometers
Beim Austritt aus der Ionenquelle ist die kinetische Energie der Ionen gleich der Beschleunigungsenergie des Feldes in der Ionenquelle. Der Ionenstrahl gelangt als nächstes in das Magnetfeld, das senkrecht zu der Bewegungsrichtung der Ionen angelegt ist. Durch die Lorentzkraft werden die Ionen auf eine Kreisbahn abgelenkt. Der Ablenkradius ist abhängig vom Masse/Ladungs-Verhältnis des Ions und lässt sich durch Gleichsetzen von Lorentzkraft und Zentrifugalkraft berechnen:
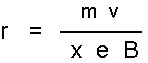
r = Ablenkradius
m = Masse des Ions
v = Geschwindigkeit des Ions
e = Ladung des Ions
B = Magnetfeldstärke
x = Anzahl der Ladungen
Aus der Gleichung ist ersichtlich, dass der Ablenkradius r vom Verhältnis m/z (mit ex = z) abhängig ist. Das Magnetfeld bewirkt aber nicht nur eine Auftrennung der Ionen nach ihrem m/z-Verhältnis, sondern auch eine Richtungsfokussierung. Selbst in einem scharf gebündelten Ionenstrahl haben nicht alle Ionen genau dieselbe Richtung. Durch den Umlauf um 180° durch das Magnetfeld wird diese Dispersion korrigiert und die Ionen mit gleichem m/z werden in einem Punkt vereinigt.
Zur Aufnahme eines Massenspektrums können wahlweise der Radius r, die Magnetfeldstärke B oder die Spannung U variiert werden. Der Radius eignet sich nicht, da dann entweder ein Detektor örtlich verschiebbar gestaltet oder mehrere Detektoren fest eingesetzt werden müssten, was für die organische Analytik zu aufwendig ist. Die Variation der Spannung ist technisch einfacher und billiger zu realisieren als die Variation der Magnetfeldstärke, liefert aber eine schlechtere Reproduzierbarkeit, weil Rückwirkungen auf die Ionenquelle und damit auf die Ionisierungswahrscheinlichkeit auftreten.
Das Auflösungsvermögen eines Magnetfeldes wird begrenzt durch die Maxwell'sche Geschwindigkeitsverteilung der Ionen. Daher lässt man den Ionenstrahl zusätzlich ein elektrisches Feld durchlaufen, dessen Feldrichtung sowohl senkrecht zum Magnetfeld als auch senkrecht zum Ionenstrahl gerichtet ist. Es findet eine Geschwindigkeitsfokussierung statt, weil die Ionen nach ihrer kinetischen Energie und unabhängig von ihrer Masse, getrennt werden. Es gilt nun:
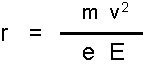
r = Ablenkradius
m = Masse des Ions
v = Geschwindigkeit des Ions
e = Ladung des Ions
E = Elektische Feldstärke
Beim Ion Trap handelt es sich um ein niederauflösendes Massenspektrometer, das vorwiegend zur GC/MS-Kopplung verwendet wird. Alle Angaben wie Spannungen oder Frequenzen beziehen sich auf das Ion Trap Massenspektrometer ITS 40 MAGNUM der Firma Finnigan MAT.
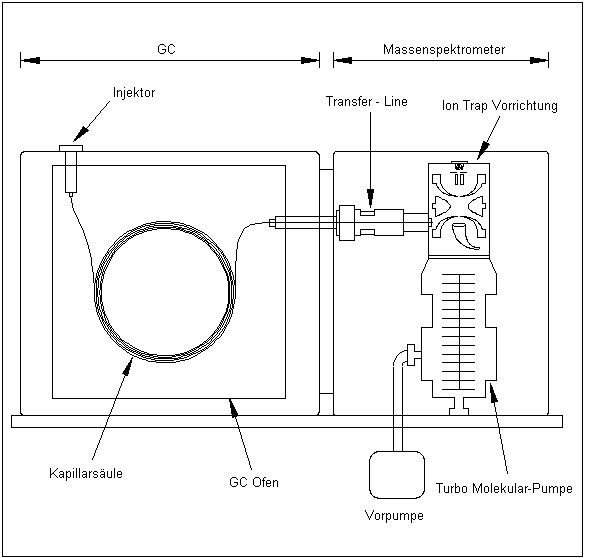
Die mittels GC getrennten Probenmoleküle gelangen mit dem Trägergasfluss über die Transferline in die Ion Trap. Hier werden diese entweder durch energiereiche Elektronen (EI) oder durch chemische Ionisation (CI) ionisiert. Die daraus resultierenden Molekülionen werden dann über ihr Masse/Ladung-Verhältnis (m/z) detektiert.
4.2 Die Bestandteile der Ion Trap
Die eigentliche Ion Trap Konstruktion besteht aus folgenden Komponenten:
- Glühelektrode
- Linsenanordnung
- Elektron Gate
- drei Ion Trap Elektroden
- Sekundärelektronenvervielfacher (SEV)
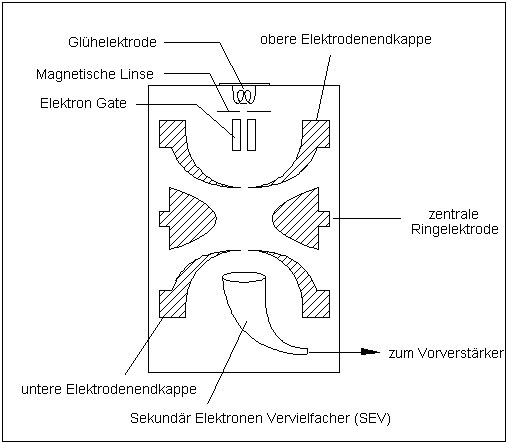
Abb. 7: Wichtige Komponenten der Ionisationskammer
Glühelektrode
Die Glühelektrodenanordnung besteht aus zwei Rhenium Drähten, die durch Anlegen eines Heizstromes von 2 bis 3 A Elektronen emittieren. In der Regel wird immer nur eine der beiden Elektroden beheizt, damit die zweite als Reserve zur Verfügung steht. Der Heizstrom wird über den Elektronenemissionsstrom geregelt. Dieser wird über das angeschlossene Datensystem eingestellt und liegt zwischen 5 und 100 µA. Hinter den Glühelektroden befindet sich ein sogenannter "Repeller" (engl. to repel = abstoßen), an dem eine Spannung von -12,5 V angelegt ist. Die emittierten Elektronen werden aufgrund des negativen Potentials abgestoßen und in Richtung der Linsenanordnung beschleunigt.
Elektronenlinse
Die Elektronenlinse befindet sich zwischen den Glühelektroden und dem Elektron Gate. Sie besteht aus einer an eine Metallplatte angeschlossene Metallröhre und reicht bis in das Elektron Gate. Die Linse fokussiert den emittierten Elektronenstrahl.
Electron Gate
Das Electron Gate besteht aus einer zylindrischen Elektrode, welche den Eintritt der Elektronen in die Ionisationskammer steuert. Werden keine Elektronen für die Ionisation benötigt, so liegt an dieser Elektrode eine Spannung von -150 V an. Hierdurch werden die Elektronen daran gehindert, in die Ionisationskammer zu gelangen. Sollen Elektronen in die Kammer gelangen, so liegt an der Elektrode eine Spannung von +180 V an. Diese kann über einen Zeitraum von 10 bis 25000 µs eingeschaltet werden. Während dieser Zeit werden die Elektronen zusätzlich beschleunigt.
Die Beschleunigungsspannung setzt sich aus mehreren Komponenten zusammen. Aus der Spannung des Repellers von -11,5 V, der an das Electron Gate angelegten Ziehspannung von +180 V und den positiven Phasen der hochfrequenten Wechselspannung (RF), welche an den eigentlichen Elektroden der Ionisationskammer anliegt. Durch die Summe dieser Spannungen treten die Elektronen mit einer Energie von 50 bis 80 eV in die Ionisationskammer ein. Hier werden dann die Probenmoleküle direkt durch Elektronenstoß (EI); oder im Falle der chemischen Ionisation (CI) die als Reagenz verwendeten Gasmoleküle ionisiert.
Ion Trap Elektroden
Die eigentliche Ion Trap Konstruktion besteht aus drei Stahlelektroden:
- der oberen Endkappe
- der zentralen Ringelektrode
- der unteren Endkappe
Alle drei Elektroden sind hyperbolisch geformt, um eine ideale Geometrie des Feldes zu erhalten, und bilden die Ionisationskammer, in der die Ionisation und die Ionen- (Massen-) trennung stattfindet. Die Ion Trap arbeitet im Gegensatz zum Quadrupole Massenspektrometer diskontinuierlich und benutzt für die Erzeugung und die Trennung der Ionen denselben Raum. Das Feld wird durch Anlegen von hochfrequenter Spannung und Gleichspannung erzeugt.
Die obere Endkappe beinhaltet die bereits beschriebene Glühelektrode sowie die Fokussiervorrichtung für den Elektronenstrahl. Durch sie treten die energiereichen Elektronen in die Kammer ein.
Die zentrale Ringelektrode ist durch Keramikstäbe isoliert aufgehängt. An ihr liegt eine Wechselspannung mit einer konstanten Frequenz von 1,05 MHz und varaibler Amplitude von 0 bis 7500 V an. Die Frequenz der Wechselspannung liegt im Radio-Frequenz Bereich, weshalb sie im Folgenden nur noch als RF Spannung bezeichnet wird.
Die untere Endkappe besitzt in ihrer Mitte sieben Löcher, über welche die gebildeten Ionen die Ionisationskammer verlassen und in den Elektronenvervielfacher gelangen.
Liegt die richtige RF Spannung an den Elektroden an, so bilden diese ein dreidimensionales, hyperbolisches, elektrisches Feld aus. Nimmt die RF Spannung zu, so werden die Flugbahnen der Ionen, in Abhängigkeit von ihrem Masse/Ladungs - Verhältnis, instabil. Ionen mit einer kleineren Masse bei gleicher Ladung werden zuerst instabil und verlassen den Ion Trap. Der SEV registriert die Ionen und liefert ein Messsignal an das angeschlossene Labordatensystem.
Während der Massenanalyse wird eine zusätzliche RF Spannung von 485 kHz zwischen den beiden Endkappen zugeschaltet. Diese Spannung verbessert das Auflösungsvermögen und die Empfindlichkeit der Analyse.
Sekundärelektronenvervielfacher (SEV)
Der SEV des Ion Trap besteht aus einem mit Bleioxid überzogenen Glastrichter (Kathode) sowie einer Metallkappe (Anode) am Ausgang des Trichters. Am oberen Ende der Kathode liegt eine negative Spannung von -800 bis -2900 V an. Das untere Ende der Kathode liegt auf Masse. Abbildung 8 zeigt schematisch die Funktionsweise des SEV.
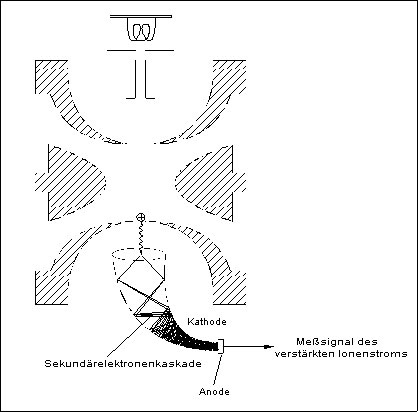
Abb. 8: Schematische Funktionsweise des Elektronenvervielfachers
Positive Ionen, welche aus der Ionisationskammer austreten, werden von der negativen Spannung der Kathode angezogen. Diese Ionen treffen mit einer ausreichend hohen Geschwindigkeit auf die innere Oberfläche der Kathode um Elektronen aus dieser herauszuschlagen. Die herausgeschleuderten Elektronen werden durch das Potentialgefälle beschleunigt und treffen erneut, als Ergebnis der gekrümmten Oberfläche, auf diese auf. Hieraus resultiert ein ständig wachsender Elektronenstrom, eine sogenannte Elektronenkaskade.
Die aus dem Ende des Elektronenvervielfachers austretenden Elektronen werden von der Anode aufgenommen und das entstehende Signal über ein Voltmeter gemessen. Das Signal ist proportional zu dem aus der Ionisationskammer austretenden Ionenstrom.
4.3 Zusammenfassung der möglichen Ion Trap Parameter
4.3.1 Steuerung der hochfrequenten Wechselspannung
Vor der Ionisation der Probenmoleküle wird die an der Ringelektrode angelegte hochfrequente Wechselspannung für eine Zeitdauer von 2 ms auf annähernd 0 V gesetzt. Diese Einstellungen sind nötig um den Hochfrequenzgenerator vor der nächsten Analyse zu stabilisieren. Während der Analyse wird die Hochfrequenzspannung auf einen bestimmten Wert, die sogenannte "Haltespannung", gesetzt. Dies ist nötig um die Ionen, die von Interesse sind, in der Analysenkammer festzuhalten (engl. trap = Falle). Nach der Ionisierungszeit wird die Spannung sehr schnell auf den Startwert hochgefahren und dann während der Analyse langsam erhöht.
Die Spannung, bei der ein Ion aus der Kammer austritt, wird als Resonanz Spannung bezeichnet. Bei einer Spannung die niedriger als die Resonanz Spannung ist, kreist das Ion auf einer stabilen Bahn und schwingt in allen drei Raumrichtungen. Nimmt die Spannung zu, wächst die Amplitude der Schwingung in vertikaler Richtung schnell an. Eine Hälfte der Ionen erreicht aufgrund dieser Bewegung die obere Elektrodenendkappe und wird neutralisiert, die andere Hälfte verläßt durch die Öffnungen in der unteren Endkappe die Ionisationskammer und trifft zu einem großen Teil auf den Elektronenvervielfacher auf.
4.3.2 Axiale Modulationsspannung
Die Anzahl der in der Ionisationskammer gehaltenen Ionen ist proportional zur Ionisationszeit und der von der Glühelektrode ausgehenden Emission an energiereichen Elektronen.
Obwohl die maximale Menge an Ionen, welche in der Kammer gehalten werden kann, bei 106 bis 107 Ionen liegt, findet bereits bei einer Anzahl von 104 bis 105 Ionen aufgrund des begrenzten Raumes eine Abstoßung zwischen den gleichgeladenen Ionen statt, was eine Verringerung des Auflösungsvermögen zur Folge hat (Raumladungseffekte). Um das zu verhindern, wird an die Endkappenelektroden eine schwache Wechselspannung, die axiale Modulationsspannung, angelegt. Diese hat eine feste Frequenz von 485 kHz. Resoniert ein Ion mit dieser Spannung, dann bewegt es sich aus dem Zentrum der Trap in einen Bereich, wo das von der RF Spannung aufgebaute Feld stärker ist. Durch die kleinere Anzahl an Ionen in diesem Bereich kommt es nur zu geringen Raumladungseffekten, was eine Verbesserung des Auflösungsvermögens zur Folge hat.
4.3.3 Gasdruck in der Ion Trap
Die Anwesenheit von Helium in der Ionisationskammer hat einen großen Einfluss auf das Auflösungsvermögen und die Empfindlichkeit des gesamten Systems. Der Gasdruck im Inneren des Trap wird zum einen über den Gasfluss aus der Kapillarsäule in die Kammer zum anderen über die Pumpleistung der Vakuumpumpe reguliert. Der Gasfluss in bzw. aus der Kammer heraus wird so eingestellt, dass ein Wert von 13,3 Pa nicht überschritten wird. Andernfalls stoßen die Ionen, bevor sie die Kammer verlassen können, mit Heliumatomen zusammen, wodurch sich ihre kinetische Energie und somit ihre Schwingungsamplitude verringert. Als Ergebnis bewegen sie sich wieder in Richtung des Zentrums anstatt die Kammer zu verlassen.
4.3.4 Elektronenstoßionisation mit Automatic Gain Control
Im Gegensatz zu einem konventionellen Massenspektrometer (z.B. Quadrupol-MS oder Sektorfeld-MS), in dem die Ionisierung und Massenanalyse kontinuierlich, aber örtlich getrennt, nämlich in der Ionenquelle und dem Quadrupolfeld bzw. Magnetfeld erfolgt, arbeitet die Ion Trap diskontinuierlich und es wird für die Erzeugung und Trennung der Ionen der gleiche Raum benutzt.
Wird mit Elektronenstoßionisation gearbeitet, wechselwirken die Probenmoleküle nach dem Eintritt in die Ion Trap mit energiereichen Elektronen, wodurch positive Ionen gebildet werden. Die drei Prozesse, die in der Trap ablaufen -Ionisation, Massenanalyse und Detektion- werden in der Regel durch die Automatic Gain Control (AGC) kontrolliert. Die AGC Scan Function kann durch ein Diagramm dargestelt werden, in dem die RF Spannung gegen die Zeit aufgetragen ist.
Die AGC Scan Function besteht aus einem Prescan und vier Scansegmenten. Die vier Scan Segmente unterteilen den gesamten aufnehmbaren Massenbereich von 10 bis 99 amu, 100 bis 249 amu, 250 bis 399 amu und 400 bis 650 amu. Ein Scan beinhaltet ein komplettes Hochfahren der RF Spannung über den Massenbereich, der vom Benutzer gewählt wurde. Die RF Spannung wird mit einer konstanten Rate von ungefähr 5600 u/s hochgefahren. Ein kompletter Scan mit dieser Rate ergibt einen Microscan.
AGC Scan Function
Die AGC Software wählt automatisch eine Ionisationszeit zwischen 10 und 25000 µs für jedes der vier Scansegmente des Microscans aus, was von der Menge der zu analysierenden Substanz in der Ion Trap abhängt. Eine Schätzung der Anzahl der in der Ion Trap gebildeten Ionen liefert ein kurzer Prescan, der aus einer 0,2 ms dauernden Ionisationsperiode besteht. Für niedrige Konzentrationen, wie z.B. die Basislinie oder kleine GC Peaks, wird eine maximale Ionisationszeit von 25000 µs ausgewählt. Wenn die Kozentration des zu analysierenden Stoffes zunimmt, nimmt die Ionisationszeit automatisch ab, um eine Überladung der Ion Trap mit Ionen zu verhindern. Die Ionensignale werden in jedem Microscan automatisch in den richtigen Maßstab gebracht, um die unterschiedlichen Ionisationszeiten wieder auszugleichen.
Dem Prescan folgt ein Hochfahren der RF Spannung, damit die Backgroundmassen bis 20 u nicht mit aufgenommen werden. Das führt dazu, dass alle Ionen mit Massen unterhalb der Backgroundmasse, d.h. solche Massen, die dem chemischen Background und den Trägergasionen entsprechen, die Trap verlassen.
Die RF Spannung wird dann auf einen Maximalwert hochgefahren und alle Ionen mit Massen oberhalb der Backgroundmasse treten aus der Ion Trap aus und werden durch den Multiplier nachgewiesen. Das Ionenstromsignal vom Multiplier ist der Totalionenstrom (TIC) des Iontrap.
Der TIC, der während des AGC Prescan gemessen wurde, wird als die AGC Peakfläche bezeichnet. Die AGC Peakfläche ist proportional zu der Anzahl der während der Prescanionisation gebildeten Ionen. Die AGC Software benutzt den gemessenen TIC als Bezug, um die Ionisierungszeit für jeden Scan in Abhängigkeit der Konzentration zu optimieren.
Dem Prescan folgen die vier Segmente des Scans. Jedes Segment kann in zwei Teile getrennt werden: eine Setup Period und eine Scanning Period. Jede Setup Period kann in die folgenden vier Schritte unterteilt werden:
- Erster Schritt: Die RF Spannung wird auf ca. 0 V gesetzt.
- Zweiter Schritt: Die RF Spannung wird auf die Haltespannung gesetzt und stabilisiert.
- Dritter Schritt: Zuerst wird das Electron Gate eingeschaltet. Energiereiche Elektronen vom erhitzten Filament werden in die Ion Trap beschleunigt, wo sie mit Probenmolekülen in Wechselwirkung treten und Ionen erzeugen. Nach der Ionisationsperiode wird das Electron Gate abgeschaltet und weitere Elektronen werden daran gehindert in die Ion Trap zu gelangen. Die neu gebildeten Probenionen können sich dann bei der Haltespannung auf ihrer Umlaufbahn stabilisieren. Die Länge dieser Stabilisationsperiode kann für maximale Spektralempfindlichkeit geändert werden. Im allgemeinen gilt, dass mit steigender Dauer die Empfindlichkeit steigt, d.h. es werden mehr Ionen gehalten.
- Vierter Schritt: Der Elektronenvervielfacher wird bereitgemacht und die RF-Spannung wird von der Haltespannung auf einen entsprechenden Wert hochgefahren, um das Durchfahren des nächsten Segments zu beginnen.
Nach der Setup Period beginnt die Scanning Period. Während dieser Zeit wird die RF Spannung mit einer konstanten Rate hochgefahren, so dass die IonTrap über einen bestimmten Massenbereich abgesucht wird. Nach Beendigung der Scanning Period wird die RF Spannung wieder auf ca. 0 V gesetzt und der Multiplier auf Standby. Der gesamte Prozess (setup und scanning) wird viermal pro Microscan wiederholt, einmal für jedes der vier Segmente.
Jedes der vier Segmente des Microscans kann unabhängig eingestellt werden, um die beste Kombination von Auflösung und Empfindlichkeit zu erreichen. Mit der AGC Scan Function ist die manuelle Regelung durch die Änderung der Ionisationszeit für jedes Segment möglich. Die Ionisationszeit kann erhöht oder erniedrigt werden durch Erhöhen oder Erniedrigen des entsprechenden Segment Tune Factors über das Instrument Control Programm.
Kontrolle der Ionenbildung
Die Ion Trap hat eine maximale Haltekapazität bei deren Überschreitung sich Auflösung und Qualität des Spektrums verschlechtern. Die AGC Software basiert auf der linearen Beziehung, die zwischen der Ionisationszeit und der Menge der entstandenen und schließlich in der Ion Trap gehaltenen Ionen besteht. Wenn die Ionisationszeit erhöht oder erniedrigt wird, wird die Zahl der erzeugten Ionen proportional dazu erhöht oder erniedrigt.
Die Ionisationszeit ist die Zeit (zwischen 10 und 25000 µs), während der die Spannung am Electron Gate positiv ist und der Elektronenstrahl vom Filament in die Ion Trap gelangen kann. Der Elektronenstrahl erzeugt Ionen durch Zusammenstoßen mit Proben- und/oder Backgroundmolekülen, die in der Ion Trap vorhanden sind. Die AGC Software sorgt für die Kontrolle der Ionisationszeit, sodass die Menge der Ionen optimal gehalten wird.
Die Anzahl der gebildeten Ionen in der Trap ist auch eine Funktion der Intensität des Elektronenstrahls. Im allgemeinen steigt mit erhöhtem Emissionsstrom vom Filament die Menge der produzierten Ionen. Die Beziehung zwischen Emissionsstrom vom Filament und dem gemessenen TIC, d.h. der Anzahl der erzeugten Ionen in der IonTrap, ist nicht ganz linear. Besonders bei einem Emissionsstrom über 40 µA und bei hohen Probenkonzentrationen ändern sich Emissionsstrom und gemessener TIC nicht linear.
Leistungsfähigkeit der Ionenspeicherung
Wenn Ionen während des Ionisationsimpulses gebildet werden, werden sie im Zentrum der Ion Trap durch die stark fokusierende Wirkung der RF Spannung, die an die Ringelektrode angelegt ist, gehalten. Diese RF Spannung gilt als Haltespannung, ab da wo sie das Feld erzeugt, das die Ionen zeitweise hält, bis diese die Ion Trap in Richtung Multiplier verlassen. Die Leistungsfähigkeit Ionen zu halten steigt bis zu einer gewissen Grenze genauso wie die angelegte RF Spannung steigt.
Die AGC Scan Function hält Ionen bei einer festen RF Spannung, bei der sich die Haltefähigkeit einpegelt, also ein Maximum an Ionen sicherstellt, die zur anschließenden Massenanalyse und Nachweis verfügbar sind. Die AGC Haltespannung ist bedeutend höher als die RF Spannung, die bei einer fest eingestellten Ionisationszeit angelegt wird; es werden nicht unbedingt mehr Ionen gehalten, aber Ionen mit höheren Massen werden besser gehalten.
Die feste RF Spannung, die an die Ringelektrode angelegt ist, hat einen weiteren Vorteil: Sie setzt die sog. "cut off"-Masse auf ungefähr 20 u. Das bedeutet, dass Ionen mit Massen < 20 nicht stabil sind und die Ion Trap verlassen, falls sie gebildet werden. Also fliegen H2O+ und H3O+ Ionen mit den Massen 18 bzw. 19 u, die aus Rückständen von Wasserdampf im Trägergas und innerhalb des Vakuumsystems entstehen, raus, sowie sie entstehen. Das schränkt die Möglichkeit ein, dass sie mit Probenionen reagieren und es können mehr Probenionen gehalten werden, was eine bessere Qualität und Empfindlichkeit zur Folge hat. Man kann jedoch trotzdem einige Signale bei diesen Massen sehen; im allgemeinen ist es effektiver, wenn ein größerer Abstand zur "cut off"-Masse besteht.
AGC off
Wenn das AGC abgeschaltet ist, wird eine feste Ionisation Time Scan Function benutzt, um die Prozesse, die in der IonTrap stattfinden, aufeinander abzustimmen. Diese Scan Function besteht aus vier Segmenten, die sich qualitativ mit denen der AGC Scan Function vergleichen lassen. Die feste Ionisation Time Scan Function enthält jedoch keinen Prescan. Also wird die Ionisationszeit für jedes Segment nicht je nach Konzentration der Probe variiert.
Bei AGC off kann manuell durch die Änderung der RF Haltespannung für jedes Segment des Microscans geregelt werden.
Ohne AGC wird gewöhnlich nur gearbeitet, wenn die interessierenden Ionen Massen unter 20 u haben. Die AGC Scan Function benutzt eine höhere RF Spannung, um Ionen zu halten, als dies bei einer festen Ionisationszeit der Fall ist. Deshalb werden bei abgeschalteter AGC auch Ionen mit Massen von 10 bis 20 u gehalten und vom Multiplier erfasst.
Target
Die AGC Software setzt die Ionisationszeit so, dass die optimale Menge an Ionen für jedes der Segmente erhalten wird und zwar entsprechend folgender Gleichung:
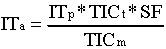
wobei:
ITa die aktuelle Ionisationszeit, die für alle Segmente in einem
4-Segment- Microscan benutzt wird;
ITp die Ionisationszeit des Prescan;
TICm die Fläche in Counts des Totalionenstroms von der Prescan-Messung;
TICt der einstellbare Targetwert des Totalionenstroms, der als Referenz zur Kalkulation der Prescan-Messung gebraucht wird;
SF ein prozentueller Anteil der aktuellen Ionisationszeit ITa, der eine individuelle Einstellung der (Ansprech-)Dauer und Auflösung für jedes Segment erlaubt.
Durch Erhöhung des Target TIC erhöht sich auch die Ionisationszeit für alle Segmente und umgekehrt. Mit dem Target TIC wird die allgemeine Auflösung über den gesamten Massenbereich (alle vier Segmente zusammen) eingestellt.
4.4 Geräteoptimierung am Ion Trap
Im Folgenden werden Möglichkeiten aufgezeigt, wie durch Geräteeinstellungen die Qualität von Messungen optimiert werden können.
Über das Instrument Control Programm können die Parameter vom Massenspektrometer eingestellt und angezeigt, die Massenspektrometerdaten direkt beobachtet und das Massenpektrometer optimal durch Set up, Tune und Kalibration eingestellt werden.
Das bedeutet, dass über den Befehl
- File der Tune File ausgewählt und die Tune Parameter geändert werden
- über Control und Set Instrument Parameters die axiale Modulationsspannung, der Emissionsstrom der Glühkathode und die Manifold Temperatur eingestellt werden
- über Control und Select Ionisationmode zwischen EI und CI gewählt wird.
Über den Befehl Auto Set Up justiert sich das Gerät so, dass die bestmögliche Kombination von Peakhöhe (Empfindlichkeit), optimaler Auflösung der Peaks und Peakform erreicht wird. Durch das Tuning soll die Anzahl der Ionen in der Ion Trap optimiert werden. Bei zu wenig Ionen leidet die Empfindlichkeit; bei zu vielen die Auflösung und die Peakform. Die zwei Hauptfaktoren, die die Ionenanzahl beeinflussen, sind die Ionisationszeit und die Amplitude der RF Spannung während der Ionisation.
Zum automatischen Tuning bei EI gehören u.a.:
- Anwesenheit von Wasser oder Luft überprüfen
- Einstellen der Multiplier Spannung
- Einstellen des Glühelektrodenstroms
- Einstellen des Target TIC
- Massenkalibration
5 Unterschiede im Auflösungsvermögen verschiedener Massenspektrometertypen
5.1 Allgemeine Definition des Auflösungsvermögens
Unter dem Begriff Auflösungsvermögen R wird im allgemeinen das Verhältnis von einer Ionenmasse m zur Massendifferenz ∆m verstanden:
R = m/∆m
∆m gibt den kleinsten Massenabstand, bei dem Massenlinien gleicher Intensität noch getrennt erscheinen. Um verschiedene Auflösungen miteinander vergleichen zu können, muss die Trennung zunächst definiert werden. In der hochauflösenden Massenspektrometrie wird normalerweise eine Auflösungsdefinition verwendet, bei der das Tal zwischen zwei gleich großen Signalen nicht mehr als 10 % der Signalhöhe ausmachen darf (Abb. 9). In der niederauflösenden Massenspektrometrie wird dagegen oft der Begriff der Peakhalbwertsbreite für m (also bei 50 % Peakhöhe) verwendet.
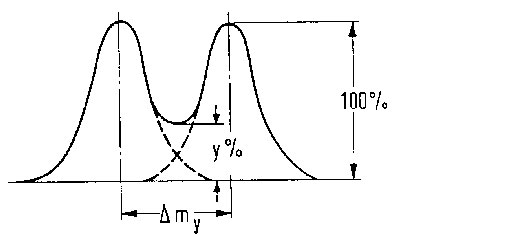
Abb. 9: %-Tal-Definition des Auflösungsvermögens
5.2 Auflösungsvermögen von Sektorfeldmassenspektrometern
Sektorfeldmassenspektrometer arbeiten über den gesamten zu detektierenden Massenbereich mit konstanter Auflösung. Der Wert R in der oben angeführten Formel ist also eine feste Größe, die entsprechend des Analysenproblems für eine Messung festgelegt wird. Daraus folgt, dass m variabel sein muss: Bei R = 2000 beispielsweise ist für die Masse 200 m = 0,1; für die Masse 200 beträgt m 1. Es wird deutlich, dass bei einer Analyse stets Peakbreiten und Intensitäten der Signale an beiden Enden des Massenbereiches betrachtet werden müssen. Bei einer hohen Auflösung wird man bei den niedrigen Massen extrem scharfe Peaks bei gleichzeitig reduzierter Intensität erhalten, mit steigender Masse nimmt die Peakbreite zu. Die Auflösung hängt im Wesentlichen von der Breite des Ionenstrahls, die durch die Fokussierung und die Öffnung des Ein- und Austrittsspaltes bestimmt wird, ab. Je enger die Spalte, desto weniger überlappen sich Ionenstrahlen unterschiedlicher Masse, und desto besser die Auflösung. Die Empfindlichkeit nimmt mit besserer Auflösung jedoch ab, weil durch den engeren Spalt geringere Intensitäten registriert werden. Die doppeltfokussierenden Massenspektrometer werden meistens "hochauflösend" (HRMS) genannt. Dies ist kein definierter Begriff. Es kann eine Auflösung bis zu 100.000 erreicht werden, was allerdings nur für Forschungszwecke sinnvoll ist.
5.3 Auflösungsvermögen von Quadrupolmassenspektrometern
Das maximale Auflösungsvermögen der Quadrupolgeräte ist abhängig von der Anzahl der Zyklen, die ein Ion im Feld durchläuft, und damit abhängig von der Länge der Quadrupolstäbe. Je länger die Stäbe (ca. 5 - 20 cm), desto höher die maximal erreichbare Auflösung. Wie schon ausgeführt, können die elektrischen Felder von Wechselspannung und Gleichspannung so überlagert werden, dass nur Ionen mit bestimmten m/e den Analysator passieren. Wie groß dieser Stabilitätsbereich ist, hängt vom Verhältnis zwischen RF-Spannung und Gleichspannung ab. Üblicherweise wird dieses Verhältnis konstant gehalten, so dass m über den ganzen Massenbereich konstant ist, während R jetzt wegen R = m/∆m eine Funktion der Masse ist. In der Regel werden die Spannungen so angelegt, dass m und (m+1) noch voneinander getrennt werden, m also =1 ist. Man spricht in diesem Fall von "Einheitsauflösung". Mit ∆m = 1 ergibt sich bei der Masse 200 eine Auflösung R = 200; bei der Masse 1000 wäre R = 1000. Die erreichbare Auflösung wird meistens in der Form: "R=A * m" angegeben. A ist eine Konstante, die normalerweise 2; 3 oder 4 beträgt, m ist die interessierende Masse. Der Massenbereich, für den Quadrupole einsetzbar sind, reicht bis ca. 1500 amu. Das Quadrupolgerät QP-5000 von Shimadzu kann für Moleküle bis ca. 700 amu eingesetzt werden. Seine Auflösung beträgt 2 m bei einer 50 %-Tal-Definition. Quadrupolgeräte werden in der Praxis wie alle anderen einfach fokussierenden Massenspektrometer "niederauflösende Massenspektrometer" genannt.
5.4 Auflösungsvermögen von Ion Trap Massenspektrometern
Da es sich bei Ion Traps auch um Quadrupolgeräte handelt, gelten die im vorigen Kapitel getroffenen Aussagen auch für diese Geräte. Bei Ion Traps kann aber die Tatsache, dass es sich um ein Speichermassenspektrometer handelt, dazu benutzt werden, die Aufenthaltsdauer im Analysator zu erhöhen und damit auch die theoretisch erreichbaren Auflösungen zu verbessern. Genauere Informationen dazu unter [1].